| Heme-proteins show various spin-states, which significantly characterize their enzymatic activity. For instance, cytochrome c' and HRP exhibit an intermediate (S=3/2) and an admixed spin-state (S=5/2, 3/2), respectively. Because the S=3/2 character is essential for the enzymes, we are constructing the corrphycene having phenyl substituents to artificially control the spin state of the metallocore. | 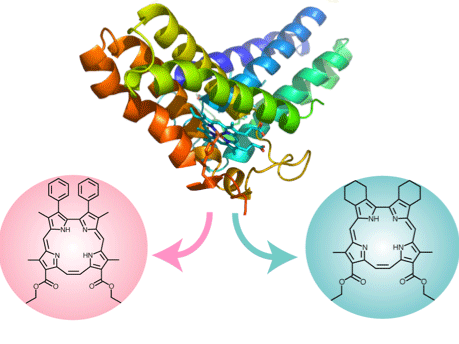 |
| Corrphycene is a kind of porphyrin isomer. No successful application of water-soluble iron corrphycene to hemoproteins has been reported. We are synthesizing a corrphycene with free acid chains to reconstitute myoglobin in aqueous medium. The functional analysis of the reconstituted myoglobin will be reported. | 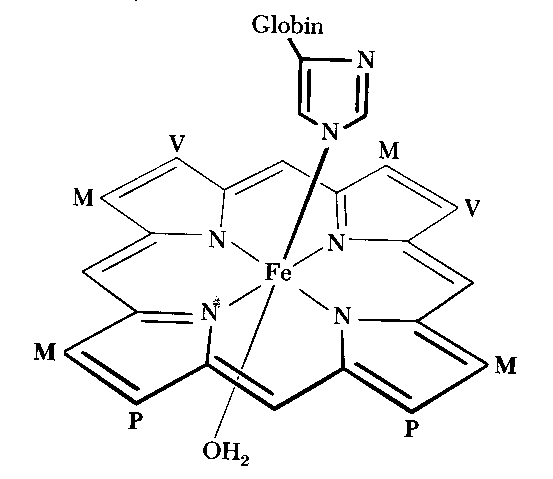 |
| We modify a part of protoporphyrin IX and synthesize mono-phenylporphyrin with carboxy groups or nitro groups. The porpose of this study is to investigate the bioinorganic characters of the synthesized porphyrins. |  |
| It have been reported that novel porphyrin derivatives or isomers are synthesized, and novel functions and characters of reconstituted heme-proteins with the porphyrin derivatives are examined. Heme is a porphyrin or porphyrin isomer in complex with Fe. In reconstituted heme-protein, native heme is replaced by modified heme. In our research, we synthesize a porphyrin isomer which has one sulfur atom and three nitrogen atoms in its center. We aim to clarify the characteristic of metal complex of the porphyrin isomer and the function of the alternate molecule. | 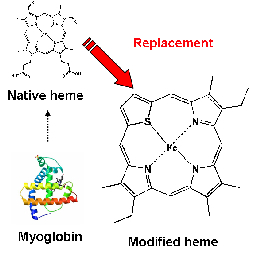 |
| when we analyze a heme-protein by 1H-NMR,it is hard to determine a character of heme-protein. Thus, we aim to be able to analyze a heme-protein, using a porphrin by 13C. First, we synthesize 13C-labeled artificial porphyrin. | 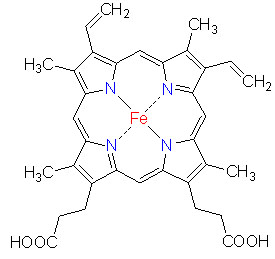 |
| HIV-1 has higher mutation rate than other viruses. Therefore, the drug-resistance against anti-HIV-1 drugs is caused more frequently. This is a serious problem of AIDS therapy. Although the amino acids related to the drug-resistance are identified, the atomistic-level resistant mechanisms are not clarified. We execute molecular dynamics simulations of the complexes of mutant proteases and inhibitors, and attempt to identify the resistant mechanism. We expect that further investigation helps not only the development of new potent HIV-1 protease inhibitors but also selection of the drugs in AIDS therapy. | 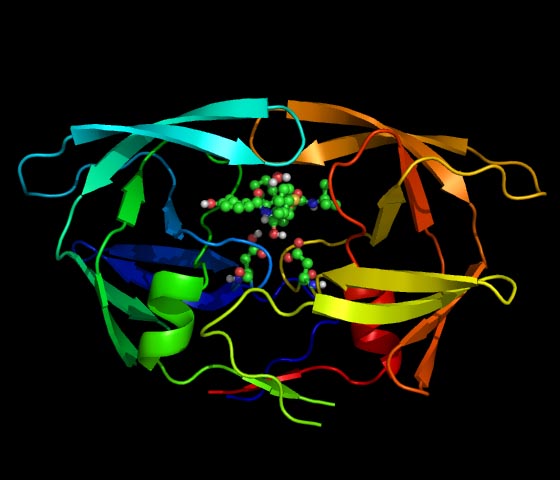 |
| The significance of Structure-Based Drug Design (SBDD) is increasing in the development of new drugs. However, in many cases when drugs are developed using SBDD, it is not clear what kind of ligands is bound to a target protein and where the binding site of ligands is. Therefore, the establishment of a computer program which correctly determines the binding site and the binding mode of ligands is essential for the success of SBDD. In this study, we will develop software program predicting the binding site, the binding mode, and the binding affinity of ligands against a target protein. | 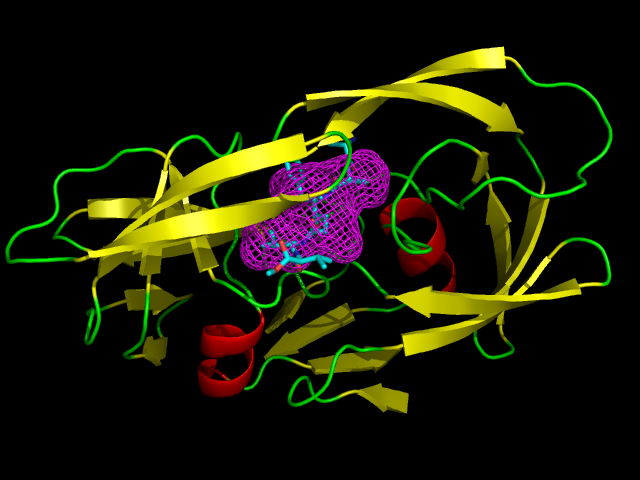 |
| Cytochrome P450 is a heme-containing enzyme that is responsible for the metabolism of a lot of known drugs. Large number of genetic polymorphisms have been described for CYP2C9 and 2D6 etc. Some of which can either result in rapid or very poor metabolism. The goal of this research is to predict metabolic ability of the enzyme protein in silico by using the method such as docking simulation or computer analysis, and to measure binding energy in vitro by the biochemical approach. | 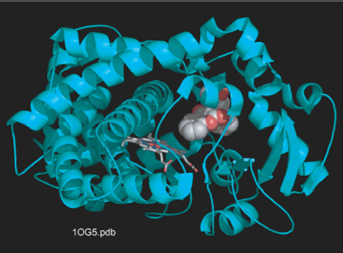 |
| When a cell is infected with virus, 8-11mer peptides (epitopes) of the virus are produced from processing by proteasome, etc. Human leukocyte Antigen (HLA) class I molecule binds to those epitopes and present them at cell surface. When the presented epitope is recognized by Cytotoxic T lymphocytes (CTLs) , the infected cell is removed. The purpose of our investigation is to assess the affinity between HLA molecule and the epitopes both in vitro and in silico. | 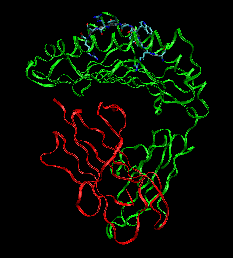 |
| HIV-1 subtype C is a majority of HIV-1 strains which are prevarent in the world, and mainly emerges in developing countries. However, no detail research have been carried out about HIV-1 subtype C, compared with subtype B variants which mainly emerge in developed countries. Hence, we plan to express clinically-derived subtype C protease and to examine thermodynamic affinity with the commercial protease inhibitors by isothermal titration calorimetry. Furthermore, we aim to execute molecular dynamics simulations to clarify drug-resistant mechanism in subtype C variants. We wish to use the results as the basic information to design novel drugs. | 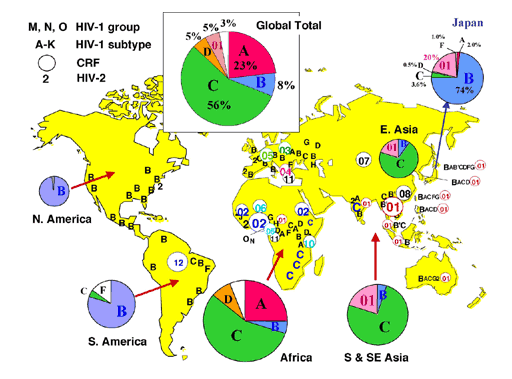 |
| Biological membranes consist of various types of lipids, and the differences of the lipid composition correspond to providing the optimal surrounding condition for specific enzymatic function. Recently, it has been revealed that cholesterols, sphingolipids, and glycolipids form more ordered, less fluid bilayers, termed microdomain, surrounded by other more fluid phospholipids. But how this heterogeneity is regulated and its functional implications remain unclear. To theoretically reveal the physical features of lipid microdomains, we have been developing new programs for modeling lipid microdomains. Utilizing these programs, we created several microdomain models, and have been performing molecular dynamics simulations of these microdomains. | 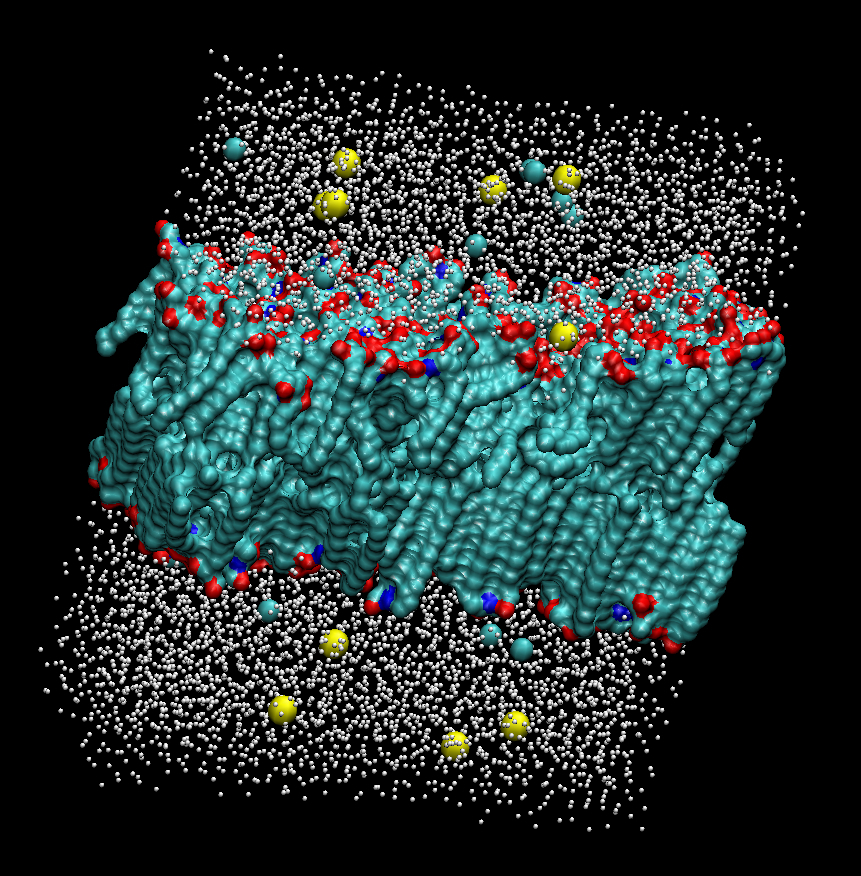 |
| For patients of AIDS, drug therapy called HAART is adapted. However, not a few numbers of cases using existing anti-drugs causes drug-resistant viruses. What we need is a novel anti-AIDS drug that acts at a different site of HIV-1. So, we focused on RNase H domain at reverse transcriptase. First, we design new compounds which hopefully inhibit the RNase H activity and perform some docking simulations. Then validate the results and carry out an organic synthesis of best scored compounds. | 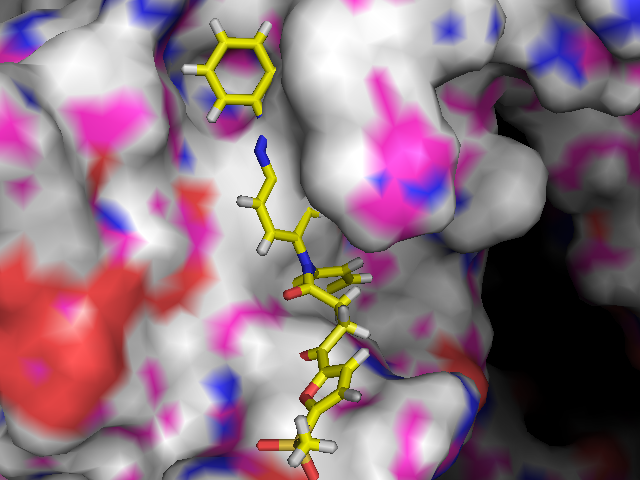 |
| Some viral proteins, such as reverse transcriptase, protease, integrase, play important role on proliferation of HIV. Hence, compounds which inhibit functions of these proteins, becomes an anti-HIV drug. An example of anti-HIV drug is inhibitors of HIV-1 protease. Darunavir, which is one of the protease inhibitor, has been recently developed. Darunavir is modified from Amprenavir, which is also the protease inhibitor, and forms stabler complex than Amprenavir. Darunavir shows high effects toward not only untreated patients but also patients in which protease confers drug resistance against some other protease inhibitors. In this study, on the basis of the previous computational study, we synthesize novel protease inhibitors modified from Lopinavir. | 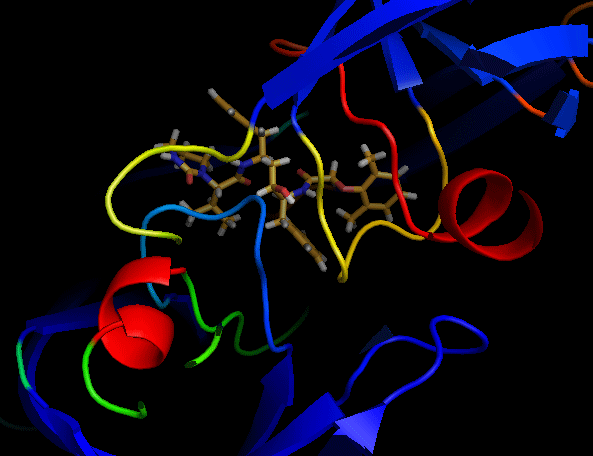 |
| A tertiary structure of a protein is one of the most important factors to design a drug. However, there are many proteins whose tertiary structure is unknown, although the amino acid sequences of them have been identified. Thus, the development of method to predict tertiary structures of target proteins on the basis of only amino acid sequences, has been noticed.One of the prediction method is ab initio method, which do not use empirical rules. In this research, we perform ab initio prediction of tertiary structures of proteins with molecular dynamics simulations. |  |
| The aim of this study is to develop antibody-drug against cancer which is highly effective and has no side effect by using computer simulation. We observe hydrogen bond, hydrophobic bond, binding energy and structural change of antibody before and after binding antigen, and then predict the amino acid sequence which enhances the antibody's affinity. Our goal is to establish the system for quick selection of antibody which has optimal amino acid sequence against antigen. | 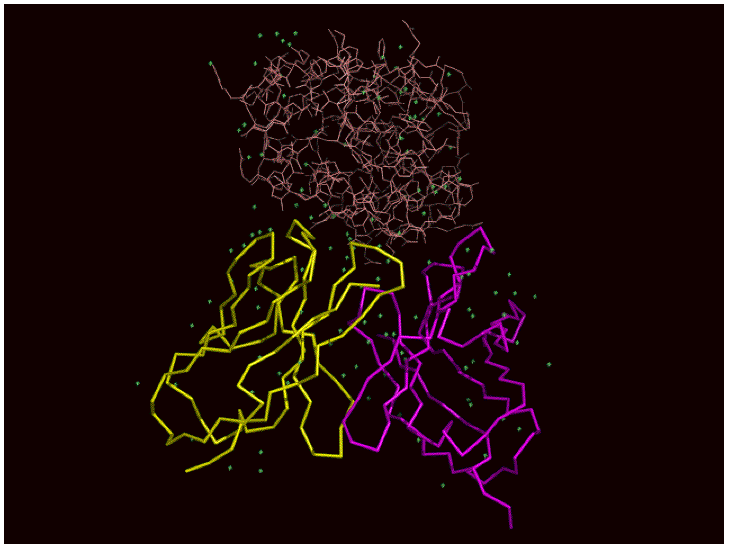 |